What is PKU (Phenylketonuria)?
Symptoms and diagnosis
Phenylketonuria, or PKU for short, is a rare autosomal recessive inborn error of phenylalanine (Phe) metabolism disorder that approximately 1:15.000 people in the world are born with.
People with PKU cannot break down an amino acid, called Phenylalanine (Phe), which consequently, accumulates in the body, becomes toxic and cause severe brain damages.
The treatment must be maintained a long-life and it consists of a low Phe diet which means a low protein diet, supplemented with Phe-free amino acid formulas to ensure normal growth and good health.
Early diagnosis and prompt intervention can enable people with PKU to grow up healthily and live a normal life, attain expected educational standards, and lead independent lives as adults.1
Phenylketonuria causes and classification
Phenylketonuria is caused by variants in the gene encoding phenylalanine hydroxylase (PAH).
PAH is a hepatic enzyme that requires the cofactor tetrahydrobiopterin (BH4) to convert Phenylalanine (Phe) to tyrosine (Tyr). A deficiency in PAH or its cofactor BH4, results in increased concentrations of Phe in the blood and toxic concentrations in the brain.
If not carefully controlled, blood Phe levels can become elevated, leading to behavioural and neurological impairments.1
Depending on the enzyme defect and the severity of the disease, different forms of PKU with different clinical phenotypes have been described. Thus, different classifications for PKU phenotypes have been established. PKU may be classified as classic PKU and milder forms of PKU (e.g., moderate PKU and mild PKU).
Definition of PKU phenotypes may be essential in establishing treatment options, e.g., new therapeutic strategies, in counseling and in prediction of the outcome, and in pregnancy.2
PKU Test: How Phenylketonuria is diagnosed?
Testing for PKU is typically done during newborn screening (NBS) about 24–72 hours after birth in the hospital, taking a blood sample from the heel of the foot and measuring blood Phe levels through a simple laboratory test. Newborn programs can vary from country to country.
What are PKU Symptoms?
Newborns with PKU initially don't have any symptoms. However, if left untreated, brain development is grossly affected, and significant intellectual disability and behavioural disturbance occur.1
Dietary treatment, particularly when maintained across neurodevelopment and well into adulthood, leads to good cognitive and psychiatric outcomes for PKU patients.3
However, it can be tough to stick to the PKU diet for life, indeed while during childhood the compliance to diet is often very high, it decreases progressively while growing up4. This is likely due to the increasing independency of patients from their families as well as to psychological and
social burdens.
Many PKU patients. initiated on dietary treatment from infancy (early-treated), who subsequently discontinued or relaxed their dietary control could experience symptoms of anxiety, depression and disturbances to cognition.5
Can PKU be prevented?
PKU is a genetic condition, and it can’t be prevented or avoided. It is inherited from parents to child in a recessive pattern, which means that to develop the disease both parents must possess a mutated version of the PAH gene. These people are called carriers.
If only one parent carries the mutated gene, the child will not develop PKU.
Even if both parents carry the mutated gene, their child still may not develop PKU, indeed in this case there also is a 25% chance that the carrier parents will both pass along the mutated gene, causing the child to have PKU.
PKU and pregnancy
Pregnancies in PKU should be planned. Women with PKU can have healthy children as long as they maintain strict adherence to their low Phe diet during their pregnancy.
Poorly controlled PKU during a pregnancy and high levels of phenylalanine can damage unborn babies, causing birth defects.
The closer to normal the mother's Phe level in her blood plasma is to normal, the less likely these complications occur, for this reason it is suggested to try to conceive once Phe levels are within the target range for pregnancy. When this is not possible, for example in case of unexpected and unplanned pregnancies, immediate referral should be made to the metabolic centre especially if phe levels are not controlled.
Additionally, pregnancy needs to be monitored very carefully even if Phe levels are under control from the beginning: at least one visit every trimester is recommended, but many clinicians may advocate more frequent follow up and the intensity of monitoring will depend on individual needs and metabolic control.1
PKU treatment
As an amino acid, Phe is a building block of proteins and is found in all proteins and most foods, therefore the treatment involves a particularly restricted and lifelong diet whose aim is to keep low Phe levels in the blood1,6. The PKU diet consists in:

Basically, no meat, fish, eggs, dairy are allowed

For many, pasta / bread / biscuits should be "special", meaning low protein

Most vegetables and fruits would need to be weighted
The main protein source comes from dedicated protein substitutes, usually based on phenylalanine-free amino acids formulas, that are essential to promote normal growth, prevent protein deficiency, provide a source of tyrosine, and help optimise blood phenylalanine control.6
Unmet needs related to traditional protein substitutes
Traditional protein substitutes contain a mixture of free amino acids and should be taken, accordingly with the European guidelines in small, frequent doses at least 3/4 times spaced evenly throughout the day.1 Nowadays there is a wide choice available of different tastes and formats, however international PKU experts suggest that improvements are still possible, in order to support PKU patients and to address some unmet needs that still exist today related to Compliance and Metabolic Management.7
1. Compliance
Growing with phenylketonuria can get tough sometimes and may become an invisible burden for people living and dealing with it.
Following the diet every day may affect PKUers’ social lives around meals and taking protein substitutes regularly may be really difficult, due to their formats and characteristics.
Compliance is affected also by the unpalatable organoleptic features of amino acids (taste, odour and persistent aftertaste in the mouth after consumption)7, deeply compromising patients’ social lives.
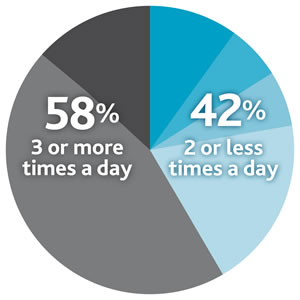
In addition, a recent study conducted across overall compliant patients showed that
frequency of amino acid mixture intake may play a tricky role in the PKU treatment, showing that 42% of PKU patients divide their total protein substitutes between ≤2 doses/day (versus the 3/4 times suggested by the European guidelines).
8
2. Metabolic Management
Another important aspect to consider within PKU treatment is related to the way amino acids are absorbed by the body after their intake protein substitutes.
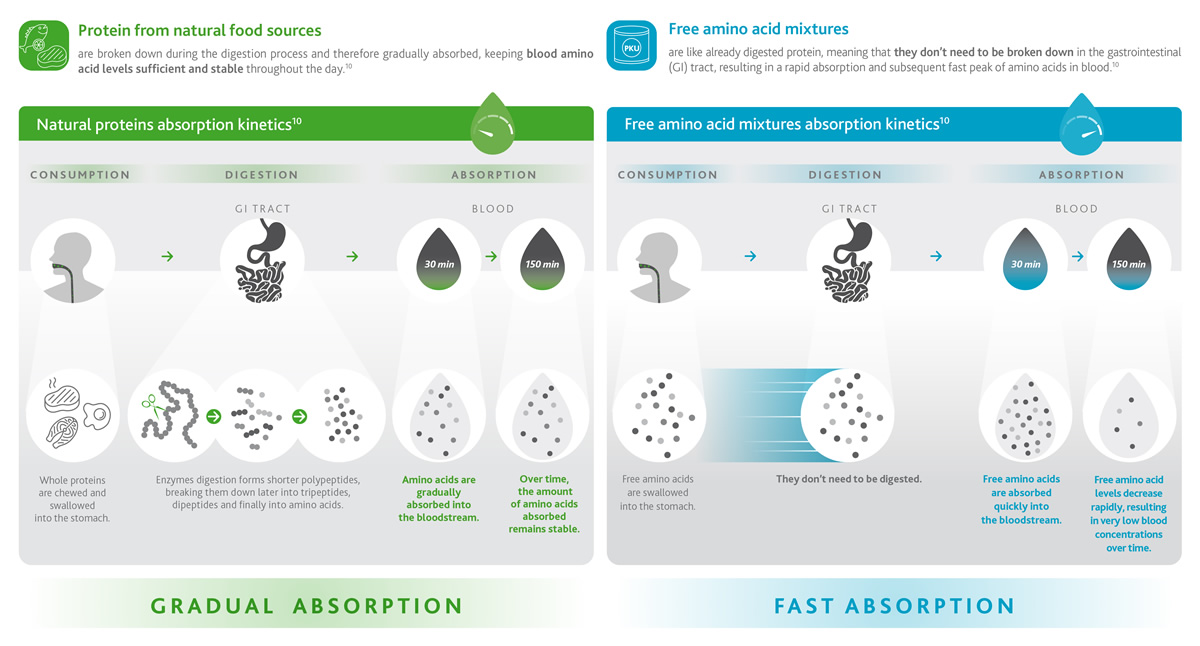
Non-PKUers would consume natural proteins within their food (such as meat, fish, dairy, eggs and so on) which are digested and absorbed gradually over time (green box), maintaining constant levels of amino acids in the bloodstream for up to 150 minutes (2.5 hours).
For PKUers protein-based food needs to be replaced by free amino acids contained in traditional protein substitutes. However, proteins and amino acids behave very differently!
Free AAs are like already digested proteins so they become directly available for absorption by the intestine. This means that a high level of amino acids is found in the blood rapidly and right after consumption of protein substitutes,9 reaching maximum peaks in only 30 minutes and drastically decreasing soon after (blue box).1,10
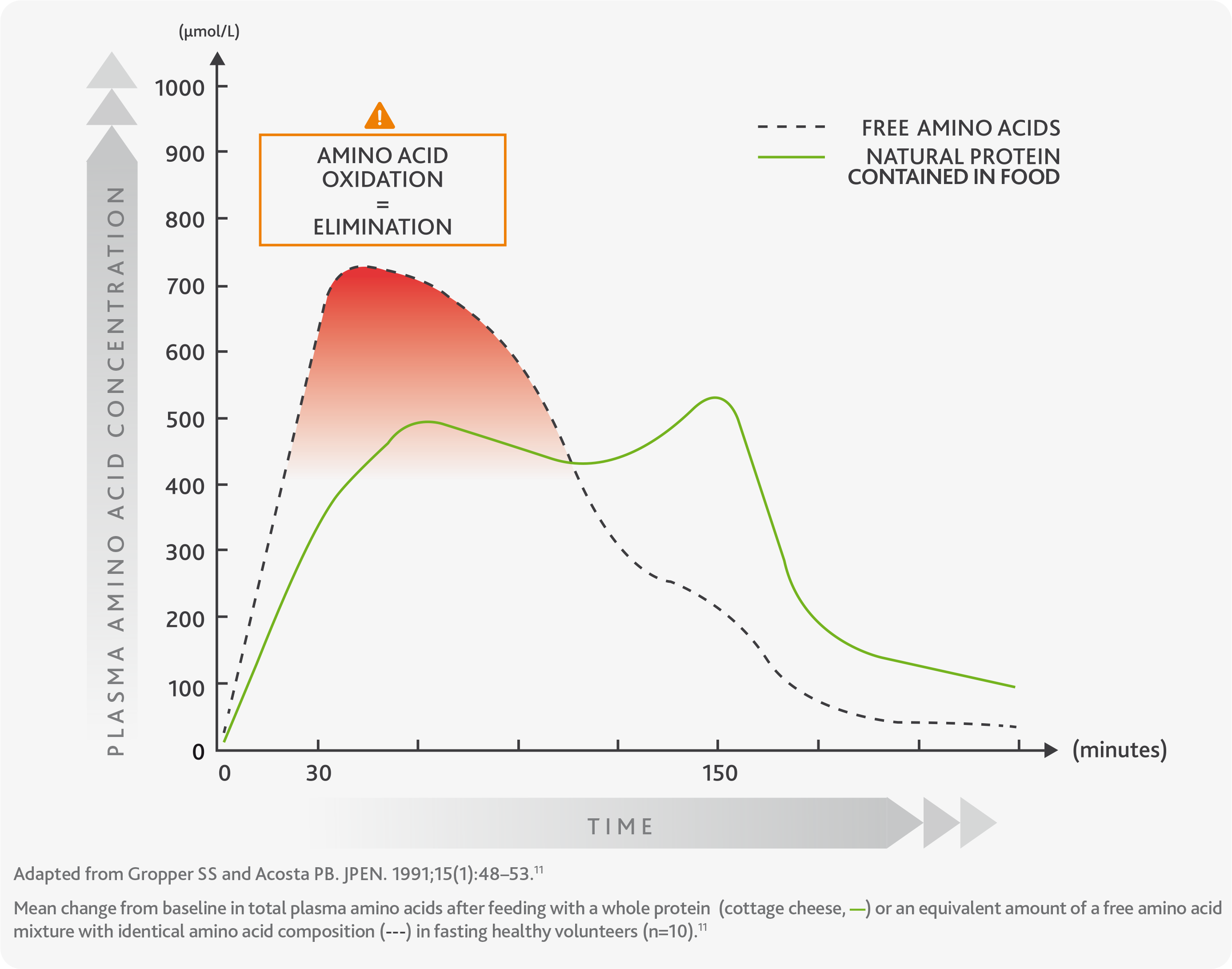
The graph shows more in detail the difference in absorption kinetics between natural protein (green line) and free amino acids (dashed line), highlighting their high waste and elimination soon after the intake (oxidation, red area) and in the so-called ‘fasting periods’, meaning portions of time during which the amount of amino acids in the body is very little.10,11
The oxidation and rapid elimination of amino acids prevents the body from using them efficiently for physiological processes such as building new proteins, having an impact on body metabolism and consequently affecting health.10,11
The ineffective absorption of amino acids and the body's inability to use them efficiently is the reason why European guidelines for PKU suggest consuming traditional protein substitutes in small and frequent doses at least 3/4 times per day.
The total protein intake should supply 40% more than the FAO/WHO/UNU safe levels of protein
1,6 in order to maintain constant levels of amino acids in the blood.
However,
this is often very difficult to accomplish and maintain, and often due to the poor biological efficacy of traditional amino acid formulas even
in adult compliant patients, an elevated risk remains of LOW mood, anxiety, and attentional disorders across the lifespan.
12
How can the PKU treatment be improved?
International experts agree that further changes are needed for protein substitutes to be improved and really make a difference in the lives of people living and dealing with PKU.
The common aim should be offering optimised taste and odour-free products, with the properties of natural protein delivering a stable chemical environment associated with better physiological function and patient tolerance7.
This means offering PKU products capable of addressing the unmet needs of compliance and metabolic control.
In order to achieve this goal and improve patients’ quality of life, APR Applied Pharma Research developed a pharmaceutical technology that for the first time was applied to a Food for Special Medical Purposes for the dietary treatment of PKU.

References:
- Van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017;12(1):162
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and
tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104
- Walter, John Η., and Fiona J. White. "Blood phenylalanine control in adolescents with phenylketonuria." International journal of adolescent medicine and health 16.1 (2004): 41-46.
- Enns,Greg et al. Molecular genetics and metabolism 101 2-3 (2010): 99-109
- Altman G, Hussain K, Green D, Strauss BJG, Wilcox G. Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet J Rare Dis. 2021 Dec 20;16(1):520
- MacDonald, A., van Wegberg, A.M.J., Ahring, K. et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare
Dis 15, 171 (2020).
- Daly A, et al. Protein substitutes in PKU; their historical evolution. Nutrients. 2021;13(2):484.
- Klimek A, Baerwald C, Schwarz M, et al. Everyday life, dietary practices, and health conditions of adult PKU patients:
a multicenter, cross-sectional study. Ann Nutr Metab 2020;76(4):251–258
- Wu G. Amino acids: metabolism functions, and nutrition. Amino Acids 2009;37(1):1–17
- MacDonald A, Singh RH, Rocha JC, van Spronsen FJ. Optimising amino acid absorption: essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr Res Rev 2019;32(1):70–78
- Gropper SS and Acosta PB. Effect of simultaneous ingestion of L-amino acids and whole protein on plasma amino acid and urea nitrogen concentrations in humans. J Parenter Enteral Nutr. 1991;15(1):48–53
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments. Front Psychiatry. 2019;10:561.